Fallvorstellung: Gentherapie bei schwerer Hämophilie A
Hämophilie A ist eine genetische Erkrankung, die X-chromosomal-rezessiv vererbt wird. 30 % der Fälle sind sporadische Erkrankungen infolge Spontanmutationen des F8-Gens auf dem X-Chromosom. Klinisch auffällig sind aufgrund des Erbgangs vor allem Männer, Frauen sind dagegen meistens Konduktorinnen (Überträgerinnen) und zeigen kaum Blutungssymptome.
DR. MED. ADRIANNA JAGIELLO

Eine Mutation im F8-Gen, das für den Faktor VIII codiert, führt zu einer verminderten bzw. fehlenden Produktion von Gerinnungsfaktor VIII. Dieser ist physiologischerweise für eine ungestörte Gerinnung notwendig. Fehlt dieser bzw. ist er nur in einer sehr niedrigen Konzentration verfügbar, kommt es zu einer Gerinnungsstörung, die klinisch schon unter der Geburt zu Blutungen führen kann. Folgende Blutungssymptome können auftreten: Nabelschnurblutungen nach Entbindung, Hämatome, Gelenk-, Muskelblutungen, Nachblutungen nach OPs oder Zahnextraktionen, Hypermenorrhoe und Epistaxis. Laborchemisch liegt eine isolierte Verlängerung der aPTT bei normalem Quick-Wert vor. Außerdem ist bei der quantitativen Bestimmung des Gerinnungsfaktors VIII ein Mangel nachweisbar. Je nach Restaktivität des Gerinnungsfaktors VIII wird die Hämophilie A in unterschiedliche Schweregrade eingeteilt. Eine Restaktivität von 5–40% spricht für eine milde (leichte), eine Restaktivität von 1–5% für eine mittelschwere (moderate) und eine Restaktivität von < 1% für eine schwere Form der Hämophilie A.
Klinisch auffällig werden vor allem Betroffene, die von einer schweren bzw. mittelschweren Form betroffen sind. Je niedriger die Restaktivität des Faktors VIII ist, desto früher und häufiger treten Blutungen auf. Betroffene, die schon aufgrund der Familienanamnese den dringenden Verdacht auf eine Hämophilie A haben, werden dementsprechend früh diagnostiziert und erhalten schon ab dem Säuglingsalter eine lebenslange Prophylaxetherapie, um insbesondere Hirn- und Gelenkblutungen zu verhindern. Als Standardtherapie erhalten die Patienten regelmäßig intravenöse Gaben von Gerinnungsfaktor VIII (2–3 Mal pro Woche) bzw. subkutan (1–4 Mal pro Monat) bispezifischen Antikörper (Emicizumab®). Die Dosierung wird im Verlauf entsprechend der Faktor VIII-Konzentration mit Bestimmung vor Faktorgabe (Talspiegel) angepasst. Diese individualisierte Therapie erfolgt lebenslang, kann eine Arthropathie jedoch nicht ganz verhindern und beeinträchtigt unter anderem hierdurch die Lebensqualität der Betroffenen.
Eine neue Möglichkeit bietet nun eine seit kurzem verfügbare gentherapeutische Therapie (z. B. mit Roctavian®), deren Ziel es ist, die Erkrankung kausal und nicht nur symptomatisch zu behandeln. Dabei wird ein vermehrungsunfähiges Adeno-assoziiertes Virus (AAV) eingesetzt, dass eine Faktor VIII-kodierende Sequenz enthält. Obligat ist das Nichtvorhandensein von neutralisierenden Antikörpern gegen den Vektor (AAV) und gegen den Gerinnungsfaktor VIII, sodass nur ein Teil der Betroffenen von der Gentherapie profitieren kann. Ca. 30 % der Patienten haben nämlich eine Adenovirusinfektion in der Vergangenheit durchgemacht und entsprechende Antikörper entwickelt, des Weiteren haben ca. 50 % der Patienten mit schwerer Hämophilie A aufgrund der Substitution von Gerinnungsfaktor VIII Antikörper gegen den Gerinnungsfaktor VIII entwickelt und kommen somit für die Gentherapie nicht in Frage.
Bei der Gentherapie werden einmalig AAV-Vektoren mit der entsprechenden genetischen Information intravenös verabreicht. Die Vektoren transportieren die Faktor VIII-kodierende Sequenz zu den Hepatozyten, die anschließend das genetische Material in einigen Leberzellen integrieren. Der Patient kann bei erfolgreicher Therapie nach einigen Wochen selbst Gerinnungsfaktor VIII in ausreichender Menge herstellen, sodass eine Substitution nicht mehr notwendig ist. Diese Therapie ermöglicht den Patienten ein Leben ohne regelmäßige intravenöse bzw. subkutane Substitution.
Im Anschluss an die Gentherapie müssen die Patienten engmaschig kontrolliert werden, da die Produktion von Gerinnungsfaktor VIII erst im Verlauf einsetzt und somit zunächst noch eine Substitutionstherapie mit Gerinnungsfaktor VIII notwendig ist. Außerdem können Nebenwirkungen, wie beispielsweise Kopf-, Gelenk-, Muskelschmerzen, Übelkeit, Fieber, grippeähnliche Symptome und der Anstieg der Transaminasen auftreten. Letzteres ist mit einer Leberentzündung assoziiert und bedarf einer vorübergehenden Immunsuppression mit Kortikoiden. Die immunsuppressive Therapie wirkt auch einem Abbau der integrierten genetischen Information in den Hepatozyten entgegen.
Aktuell geht man davon aus, dass die Patienten einige Jahre Gerinnungsfaktor VIII in ausreichender Menge selbst produzieren werden können. Diese Fähigkeit wird laut den aktuell vorliegenden Studien jedoch im Lauf der Jahre verloren gehen, sodass die Patienten dann wieder auf eine Standardtherapie angewiesen sein werden. Eine erneute Gentherapie ist aktuell noch nicht möglich, da durch den Kontakt des Körpers mit dem AAV-Vektor entsprechende Antikörper gebildet werden.
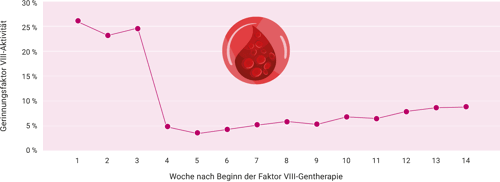
Die Gentherapie erfolgt in entsprechend zertifizierten Hämopholiezentren in Deutschland bisher nur bei einigen Patienten. Im Labor 28 konnten wir nun erstmalig den Verlauf des Gerinnungsfaktor VIII-Aktivitätsanstiegs nach einer erfolgten Gentherapie im Rahmen einer schweren Hämophilie A mitverfolgen. Der Patient hatte in den ersten drei Wochen nach Beginn der Gentherapie noch Gerinnungsfaktor VIII erhalten (FVIII-Werte um ca. 25 %). In der vierten Woche wurde die Substitution abgesetzt. Ab der ca. fünften Woche konnte man dann labordiagnostisch einen leichten Anstieg des Gerinnungsfaktors VIII beobachten. Ein Anstieg der Transaminasen ist in den ersten Monaten nach erfolgter Gentherapie nicht aufgetreten.